About Us
We utilize a hierarchical approach building on density functional theory (DFT), wave function methods(WFT), molecular dynamics (MD), and kinetic Monte Carlo (KMC) simulation to:
Study
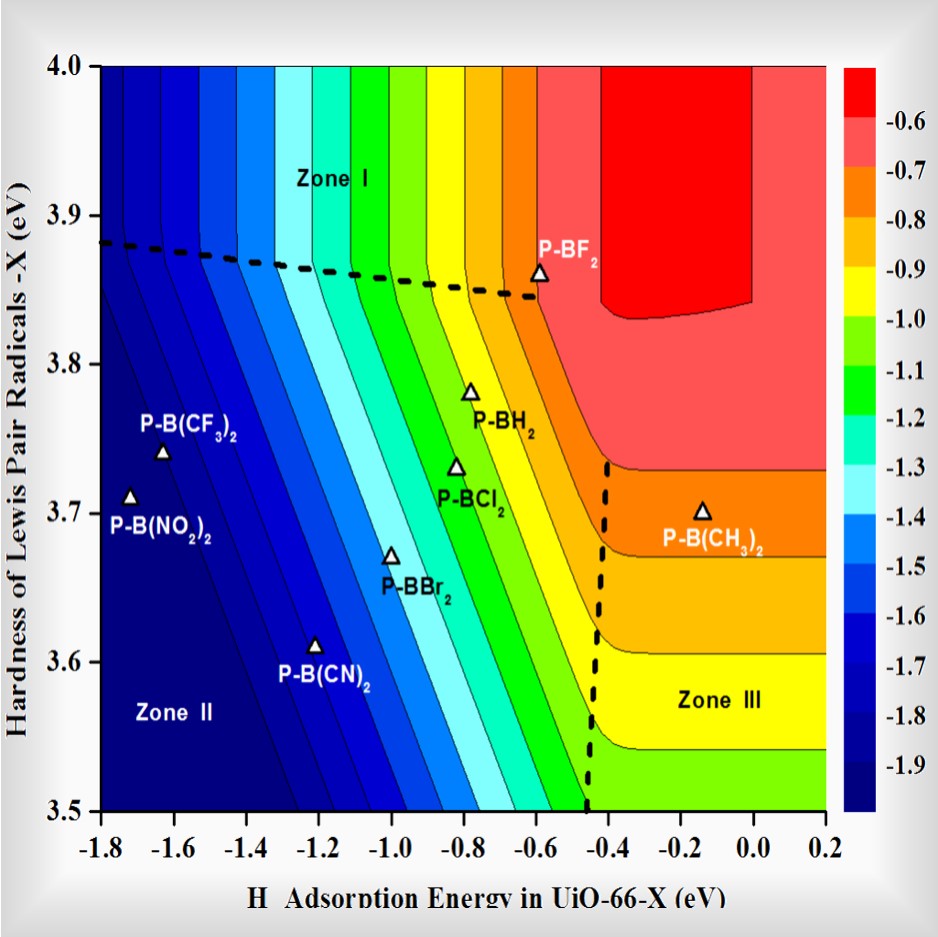
structures, properties, mechanisms, thermodynamics and kinetics of reactions for a wide range of materials including metals, metal oxides, metal complexes, zeolites, metal–organic frameworks and covalent-organic frameworks at the molecular level.
Explore
structure–function relationships to identify descriptors for large-scale computational screening, and further guide experiments to tune and improve existing materials and discover new materials.
Design
novel and multi-functional materials and efficient catalysts for sustainable energy conversion and storage.
Research Interests
Heterogeneous Catalysis
Homogeneous Catalysis
Solids

Porous Materials
Complexes
Electronic Properties
Thermaldyanmics & Kinetics